
ARTICLE SUMMARY:
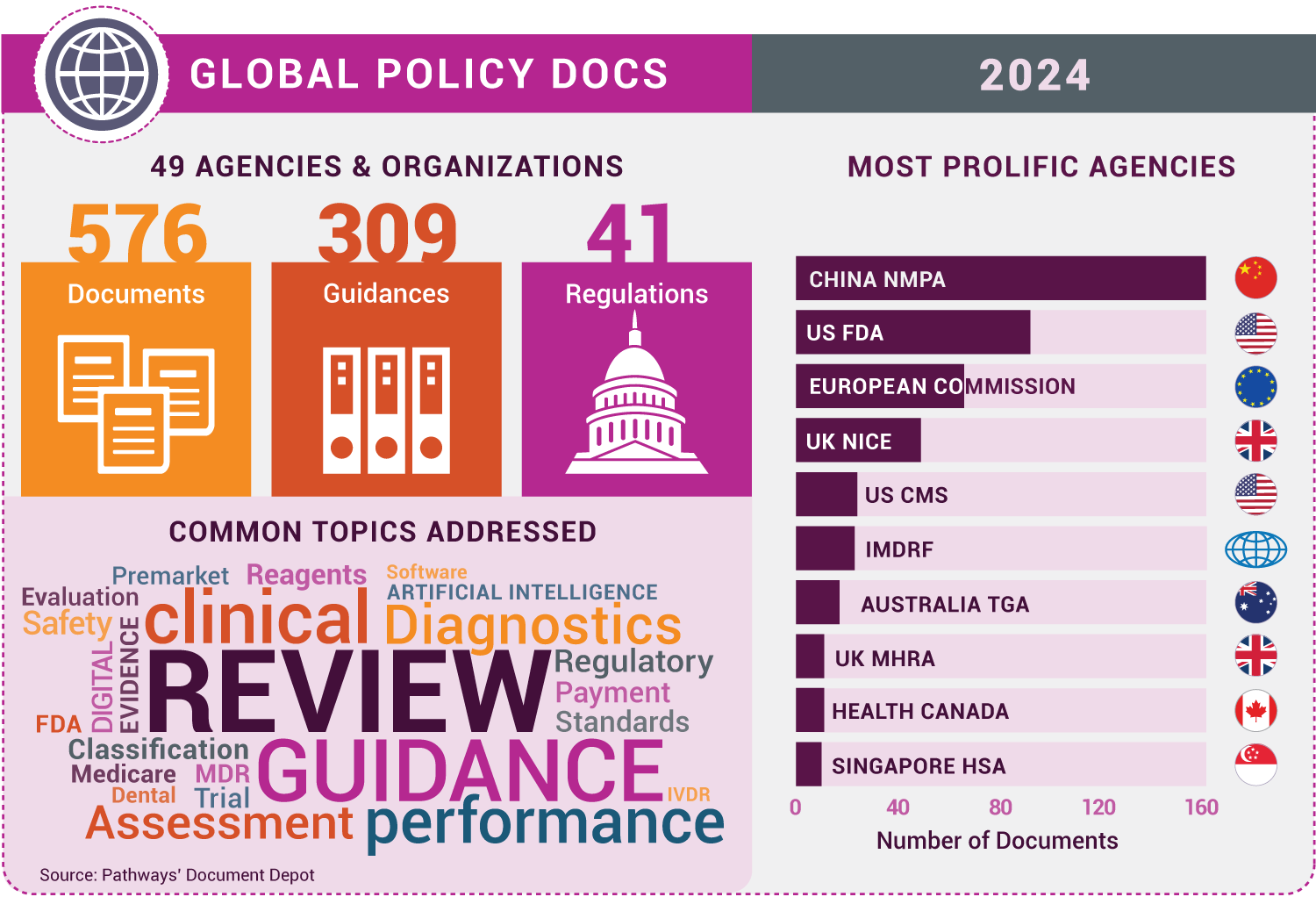
More than 500 medtech policy missives were recorded in Pathways’ Document Depot in 2024, coming from every continent except Antarctica. Here, with honorable mentions, are our top 10 documents of the year, countdown style.
[Editor’s note: This column highlights findings from Pathways’ Document Depot, a database of global medtech regulatory and policy official documents, including rules, guidance documents, memos, white papers, and more from national authorities, nongovernmental groups, and global organizations.]
Top 10 lists are inherently subjective. At their best, they generate debate about what made it on the list, and what was left off. We compiled our top 10 2024 “Docs of the Year” in that spirit. It’s an attempt to reflect regulations, guidance documents, and other policy items that we believe have a significant impact on the largest proportion of industry, that touch on weighty themes in medtech policymaking, or that simply prompt important conversations about regulation and reimbursement in the sector.
To cast a wider net, for each entry, we identified a related document as an honorable mention, and we’ve included an infographic providing a snapshot of the most active agencies and key themes in 2024 global medtech policymaking. Enjoy!
10. Laboratory Developed Tests (US FDA, April 2024)
If we were ranking documents based on the level of controversy they produce, FDA’s LDT regulation might be at the very top of this list. The rule is FDA’s attempt to end the decades-long debate about whether tests developed and performed by the same laboratory (i.e., LDTs) are subject to the agency’s medical device regulations. It lays out a four-year phase-in to definitively bring LDTs (aside from the array of grandfathered and exempted tests) into the regulatory fold, starting with recall, adverse event, and complaint reporting requirements this May. The laboratory sector fervently opposes the rule and has filed lawsuits, making clear that the LDT debate is, in fact, not settled yet. For the broader IVD/medical device industry, this is mostly a peripheral issue, but it matters primarily for two reasons: 1) the rule, if it survives court challenges, would create a more level regulatory playing field between IVD kits and LDTs, and 2) medtech groups hope opposition to the rule will eventually prompt Congress to finally pass comprehensive FDA diagnostics regulation.
Honorable mentionVerification of Manufactured Class D IVDs by Notified Bodies - MDCG 2022-3 rev.1 (European Commission, December 2024): Across the Atlantic, under the EU IVD Regulation (IVDR), high-risk diagnostics must undergo specialized batch testing and product sampling conducted by designated reference laboratories to support certification. That program was delayed in getting off the ground, but it is now finally operating for some IVD categories and this updated guidance provides the details. |
9. Clinical Evaluation of Orphan Medical Devices - MDCG 2024-10 (European Commission, June 2024)
The EU Medical Device Coordination Group has issued about 140 guidance documents and position papers tied to the Medical Device and IVD Regulations (MDR/IVDR) since 2018, but this one from June stands out. Rather than spotlight specific provisions of the MDR/IVDR and explain how notified bodies and manufacturers should implement them, as is ostensibly the case for most guidances, this one defines a product category that is not mentioned in the MDR (orphan devices) and, in the name of patient access, lays out a prospective streamlined clinical path to certification for these devices that wouldn’t normally meet the regulation’s requirements. The guidance comes in response to years of complaints that the strict MDR requirements don’t consider the challenges of products targeting patients struggling with rare conditions, and it is linked to a new EU pilot program directing Expert Panels to select developers of prospective orphan devices. How often the guidance and pilot are put to use remains to be seen, but it is almost certainly a preamble to more expansive legislative reforms to the MDR over the next few years that are expected to include a special orphan device pathway.
Honorable mentionGuideline for the Design and Statistical Analysis of Medical Device Real-World Studies for Regulatory Review (China National Medical Products Administration, January 2024): One promising evidence-generation tool cited in Europe for orphan devices is real-world data. China, meanwhile, has been making significant strides in the application of real-world data to support new device authorizations in the country. This January guidance from NMPA represents important progress in that effort, and, along with US FDA’s currently-being-updated real-world evidence guidance, should be a go-to reference for device firms looking to leverage real-world data sources to facilitate global market access. |
8. Diversity Action Plans to Improve Enrollment of Participants from Underrepresented Populations in Clinical Studies (US FDA, June 2024)
Congress passed legislation in 2022 mandating device and drug firms to submit “diversity action plans” for FDA trials, and this draft guidance is FDA’s attempt to spell out how companies should comply. It represents a major new required element of investigational device exemption and other premarket submission reviews, but companies say the draft doesn’t answer every important question. Ultimately, the diversity action plan requirement won’t kick in until six months after a final guidance is published, and some experts don’t expect FDA under the incoming Trump administration to finalize the guidance in a timely manner.
Honorable mentionHealth Equity for Medical Devices (US FDA, August 2024): Even if the new FDA leadership doesn’t act with haste to implement the diversity action plan requirement, CDRH has made clear that improving health equity in clinical studies of medical devices is one of its top priorities. This discussion paper lays out in more detail how it believes device firms should accomplish this goal. |
7. National Emission Standards for Hazardous Air Pollutants: Ethylene Oxide Emissions Standards for Sterilization Facilities Residual Risk and Technology Review (US Environmental Protection Agency, April 2024)
A 2023 proposed rule for instituting new emissions limits for EtO gas—the most commonly used sterilizer for medical devices—had industry executives sweating the prospect of massive product shortages. EPA modified some key details in this final rule published in April that make the restrictions and transitions times more reasonable from an industry perspective. Nonetheless, the rule is triggering significant infrastructure updates by manufacturers and it has catalyzed a major initiative by FDA to facilitate an industry-wide shift to reduced reliance on EtO.
Honorable mentionTransitional Enforcement Policy for Ethylene Oxide Sterilization Facility Changes for Class III Devices (US FDA, November 2024): The latest output from FDA’s EtO efforts is this direct-to-final guidance offering a streamlined approach for manufacturers responding to the EPA rule to move sterilization facilities for certain devices before winning FDA authorization for the switch. |
6. Quality System Regulation Amendments (US FDA, January 2024)
This was a major, long-time-coming accomplishment by FDA to fashion its quality management system regulations to be much closer to the global standard (ISO 13485) that most other countries use. The reality is FDA’s system, tied to US law, still has some idiosyncrasies that companies need to keep in mind. But this regulation represents an important advancement in the broader global project to harmonize medical device requirements.
Honorable mentionNormative Instruction 290 (Brazil ANVISA, April 2024): A growing focus by regulators working on harmonization is the concept of “reliance,” where a country’s authority directly leverages product decisions by another. This regulation from ANVISA is a great example, spelling out a streamlined premarket review procedure in Brazil relying on authorizations from trusted authorities in the US, Australia, Canada, and Japan. |
5. Regulation (EU) 2024/1860 (European Commission, July 2024)
Industry and government officials are ramping up efforts to make some comprehensive reforms to the MDR and IVDR, but in the meantime EU lawmakers last year were able to enact more incremental, but still pretty significant, updates to the regulation. This mid-year legislation gives manufacturers and notified bodies more time to transition legacy diagnostics to the IVD Regulation while incentivizing companies not to delay making submissions. It includes a change that will allow the Commission to finally start requiring companies over the next year or so to register in the new EUDAMED database. And it creates a new requirement (effective January 10) for companies to disclose anticipated product shortages.
Honorable mentionQ&A: Obligation to Inform in Case of Interruption or Discontinuation of Supply of Certain Medical Devices and In Vitro Diagnostic Medical Devices (European Commission, December 2024): The new mandate for companies to report anticipated temporary or permanent market withdrawals of devices that could impact public health is the most significant in the short term. This Q&A from the Commission attempts to walk firms through the ins and outs of exactly when and how they are supposed to abide by this complicated new law. |
4. CY 2025 Payment Policies Under the Physician Fee Schedule and Other Changes to Part B Payment and Coverage Policies (US CMS, November 2024)
Digital tools have thrust their way into the healthcare system, and the system is playing catch up. One big challenge is reimbursement. Established insurance systems, in particular Medicare, were not designed with the notion of nonphysical products like software serving a core therapeutic or diagnostic function. That’s why this year’s Medicare Physician Fee Schedule rule is significant: for the first time, it establishes a route for doctors to bill Medicare for stand-alone software (not embedded in a specific device), specifically FDA-authorized digital therapeutics for mental health.
Honorable mentionAct to Accelerate the Digitalisation of the Healthcare System (Digital Act, DigiG; Germany Ministry of Health, March 2024): The pioneer in health software reimbursement is Germany, which established its specialized Digital Health Applications (DiGA) reimbursement review pathway in a 2019 law. In 2024, lawmakers enacted a reform that expands the program to higher-risk apps (up to Class IIb) and institutes performance-based pricing. |
3. Transitional Coverage for Emerging Technologies (US CMS, August 2024)
On the one hand, this final notice establishing CMS’s new TCET program fell well short of the device industry’s expectations and priorities for a new streamlined Medicare coverage pathway for innovative new devices. And because the agency issued it as a notice rather than a regulation it will technically be easier for an incoming Trump administration CMS to drop it and pursue new directions. On the other hand, the TCET notice lays out a path that select FDA Breakthrough Devices can ( and are starting to) practically apply for quicker coverage, and it is the central spoke to CMS’ broader plans to be more transparent about its clinical evidence expectations for new technologies. Further, the fact that device firms and key lawmakers on Capitol Hill perceive TCET as insufficient is a primary driver for legislation that would establish a novel, more automatic coverage path for new medtech, which is the industry’s top legislative priority in 2025.
Honorable mentionBuilding an Integrated, Rules-Based Medical Technology (Medtech) Pathway: Engagement on Proposals (NHS England and the National Institute for Health and Care Excellence, May 2024): Better integrating regulatory review, technology assessment, and reimbursement procedures to speed market access for new technologies is not a challenge limited to the US. The UK health agencies, including NICE, tend to be active experimenters on this front, and this May consultation proposing a new medtech-focused market access route is one to watch. |
2. Medical Device Management Law of the People's Republic of China (Draft for Soliciting Comments) (China National Medical Products Administration, August 2024)
China holds the third-largest share of the global medical device market, after the US and the EU, and its regulator, the National Medical Products Administration, remains extremely active, as the perennial, most prolific global medtech agency tracked by Pathways’ Document Depot (see infographic). With this proposal from August, it is looking to take things to the next level by, for the first time, embedding its device framework into law. (It’s currently only defined in regulation.) How the details are worked out in a final package will be very important to determining the long-term accessibility of China’s market to new medtech innovations from different parts of the world.
Honorable mentionAnnouncement on Standardizing the Classification and Definition of Medical Devices (China NMPA, May 2024): A significant proportion of the 161 NMPA documents recorded for 2024 are device-specific technical review guidelines. But there are also several dozen more crosscutting documents that contain important updates for how medtech firms must approach the regulator of this massive market. This item from May is noteworthy in its effort to update and clarify procedures for seeking risk classification designations needed to identify the regulatory pathway for a new device. |
1. Artificial Intelligence Act: Regulation (EU) 2024/1689 (European Commission, July 2024)
The breakneck evolution of AI is perhaps the most significant technological trend for all of society, and the EU AI Act, which is the first comprehensive legal framework in the world governing development and deployment of AI, is our pick for the 2024 top document of the year. The new law goes well beyond medtech, but its implications for the increasingly AI-centric device and diagnostic sectors are substantial. AI medical device manufactures will be held to higher standards in addressing things like system bias and trustworthiness, while they, along with notified bodies and government officials, will be spending the next several years working out exactly how the new AI Act conformity assessment requirements will practically fit with the already extensive MDR and IVDR procedures. Because it is a first, the AI Act is also expected to be highly influential on the oversight of AI technology worldwide.
Honorable mentionMarketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence-Enabled Device Software Functions (US FDA, December 2024): While Europe is the first to create a comprehensive legal framework, FDA has pioneered more technical regulatory considerations for AI medtech. In particular, the agency’s conception of “predetermined change control plans,” detailed in this highly anticipated guidance, offers a novel approach to incorporate rapid iteration of AI/ML-based products into the established device review system. The guidance is expected to be influential, both within FDA (the agency has also issued a draft guidance outlining the PCCP approach for devices beyond the AI/ML realm) and globally. It bears mentioning that agencies from Australia, Taiwan, Hong Kong, South Korea, and Singapore, as well as notified bodies in Europe, issued practical guides on procedures and expectations for AI devices in 2024. There will be a lot more to come on the advancement of AI medtech oversight (at the start of 2025, for instance, FDA an even more comprehensive draft guidance on AI lifecycle management), and PCCPs are likely to play a central role. |
